The research program in the laboratory of Professor Nguyen lies at the crossroads of organic chemistry, chemical biology, organocatalysis, and transition-metal catalysis. The broad goals of our group are (1) to design and synthesize sulfated oligosaccharides and multivalent carbohydrate molecules (glycopolymers) as potent inhibitors of heparanase for treatment of cancers; (2) to design and synthesize heparan sulfate-like oligosaccharides and glycopolymers to inhibit SARS-CoV-2 virus-cell entry, thereby inhibiting progeny virus release from infected cell and spread; (3) to study saponin molecules as novel vaccine adjuvants for use in humans to combat a range of infectious diseases; (4) to develop carbohydrate molecules to control of hyperphosphorylated tau-mediated cell dysfunction and death; (5) to develop catalytic stereoselective glycosylation mediated by organocatalysts for the synthesis of bioactive oligosaccharides and glycopeptides; and (6) to investigate rhodium/iridium-catalyzed asymmetric allylic fluorination and amination as well as regioselective radio-fluorination for potential use as PET radiotracers. Overall, all of the research projects are target candidates for therapeutic drugs and PET radiotracers.
DISCOVERY OF SULFATED GLYCANS TO ATTENUATE THE COMBINED RISK OF COVID-19 AND DIABETES (Collaborators: Professor Dana Wolf and Professor Amos Panet – Hadassah Medical School)
SARS-CoV-2, the virus causing the COVID-19 pandemic, continues to spread worldwide at an unprecedented rate. SARS-CoV-2 infects the nasal epithelium, lung parenchyma, gut enterocytes, and endothelial cells. Individuals with pre-existing conditions have a higher risk of developing more severe COVID-19 associated with a higher rate of mortality. About 30% of COVID-19 deaths were associated with individual with diabetes. Further, the CDC reported that diabetes is an underlying condition for about 4 in 10 patients. Moreover, COVID-19 individuals with diabetes often develop acute metabolic complications such as diabetic ketoacidosis and hyperglycemia. Although the mechanisms underlying the link between SARS-CoV-2 and diabetes remain unclear, it is likely to involve ACE2 receptors, expressed in key metabolic organs, including the kidneys and pancreas. The SARS-CoV-2 tropism for beta cells could cause acute impairment of insulin secretion and b cell destruction, resulting in development of diabetic ketoacidosis or hyperglycaemia. Individuals with diabetes are in a chronic inflammatory state, setting them up for a more severe inflammatory response to COVID-19, which culminates in a “cytokine storm,” the prominent clinical feature of severe COVID-19 infection. COVID-19 individuals with diabetes exhibit higher levels of inflammatory biomarkers (C-reactive protein, serum ferritin) and cytokines (i.e., IL-6), and a higher erythrocyte sedimentation rate than those without diabetes. Simultaneous control of the diabetic state, SARS-CoV-2 infection, and cytokine storm is critical for the improved outcome of diabetic patients with COVID-19.
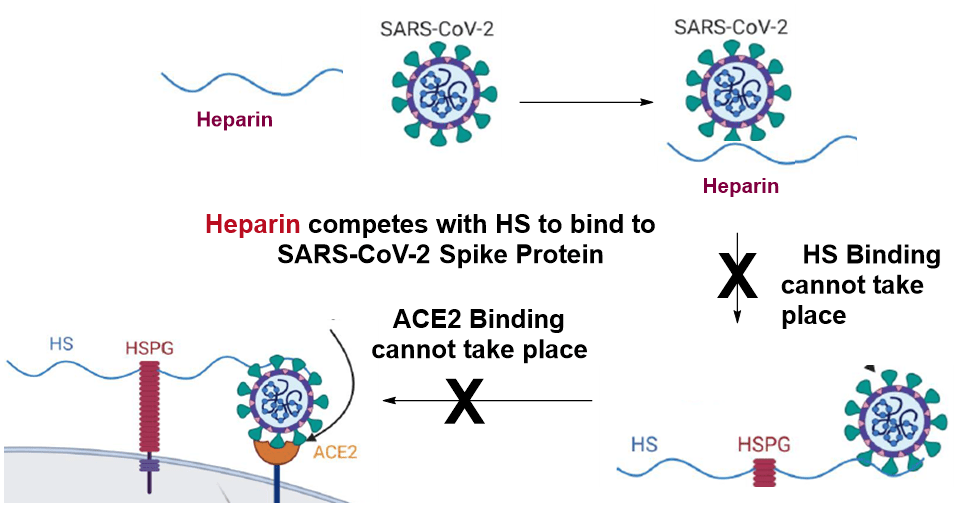
The negatively charged heparan sulfate (HS) chains interact with viral surface proteins including the spike S protein of CoV-2 and serve as co-receptors for several viruses. Consequently, HS serve as the first point of contact between target cells and many human viruses (e.g. dengue, hepatitis C, HIV, papilloma, herpes), including human coronaviruses. Coronavirus entry into host cells is mediated by its viral surface spike (S) glycoprotein that contains two subunits (S1 and S2). The S1 unit has a receptor binding domain (RBD) that binds to human angiotensin-converting enzyme 2 (ACE2) receptor to enter host cells. Heparin, low molecular weight heparin (LMWH), and some HS-mimicking compounds compete with binding of COV-2 to ACE2 receptor, indicating that HS serves as a co-receptor for ACE2 in endocytosis-mediated coronavirus entry. Our goal is to develop HS-mimicking glycans, with minimal cross bioactivity and side effects, as therapeutic intervention for diabetic individuals with COVID-19. Our work is significant because HS mimetics are regarded as promising candidates given their dual capacity to. (i) inhibit heparanase enzymatic activity and thereby protect beta cells from destruction and attenuate virus detachment/release and spread, (ii) compete with HS and thus inhibit virus-cell adhesion and entry, and (iii) reduce inflammation and cytokine storm.
OLIGOSACCHARIDE AND GLYCOPOLYMER INHIBITION OF HEPARANASE (Collaborator: Professor Israel Vlodavsky at Technion – Israel Institute of Technology)
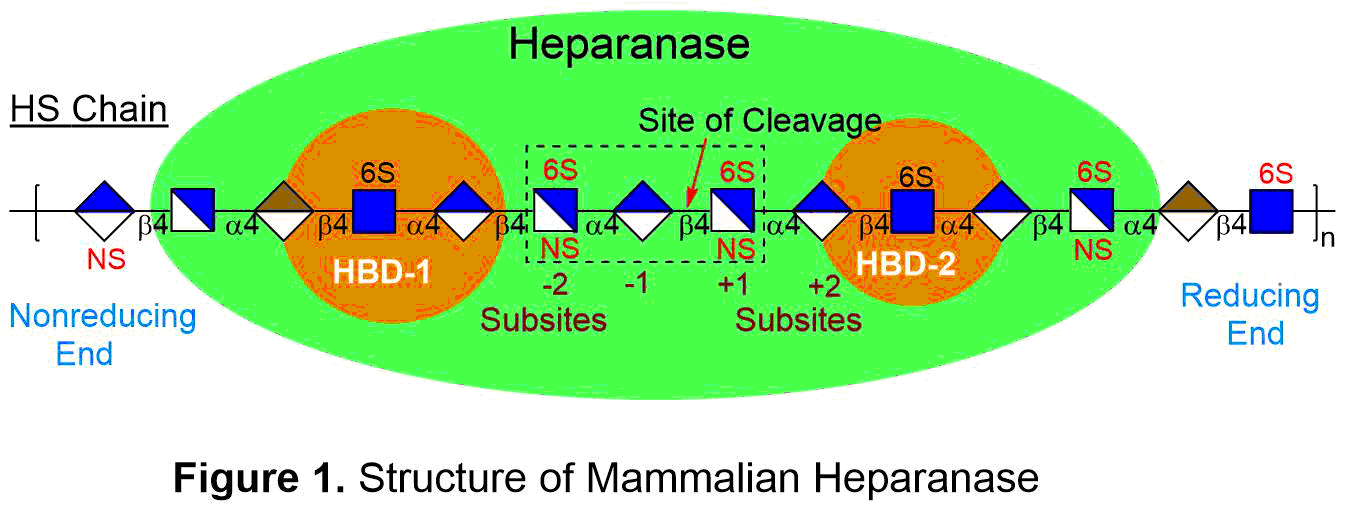
Heparanase is an endo-beta-D-glucuronidase that cleaves heparan sulfate (HS) polysaccharide chains in the extracellular matrix (ECM) and the cell surface basement membrane (BM). This enzyme is regarded as a regulator of aggressive tumor behavior as clinical studies have shown that raised heparanase levels correlated with increased tumor size, amplified tumor angiogenesis, enhanced metastasis, and poor patient prognosis. This enzyme hydrolyzes GlcAbeta(1,4)GlcNS glycosidic bonds (see Figure 1) of the HS chains releasing sequestered pools of HS oligosaccharide-binding growth factors for signaling activation, which promote tumor angiogenesis and growth. Cleavage of HS chains also degrades the structural integrity of the BM and the ECM, permitting migration of malignant cells into the blood stream, promoting cancer metastasis. Upregulation of heparanase is ubiquitous amongst all types of cancers.
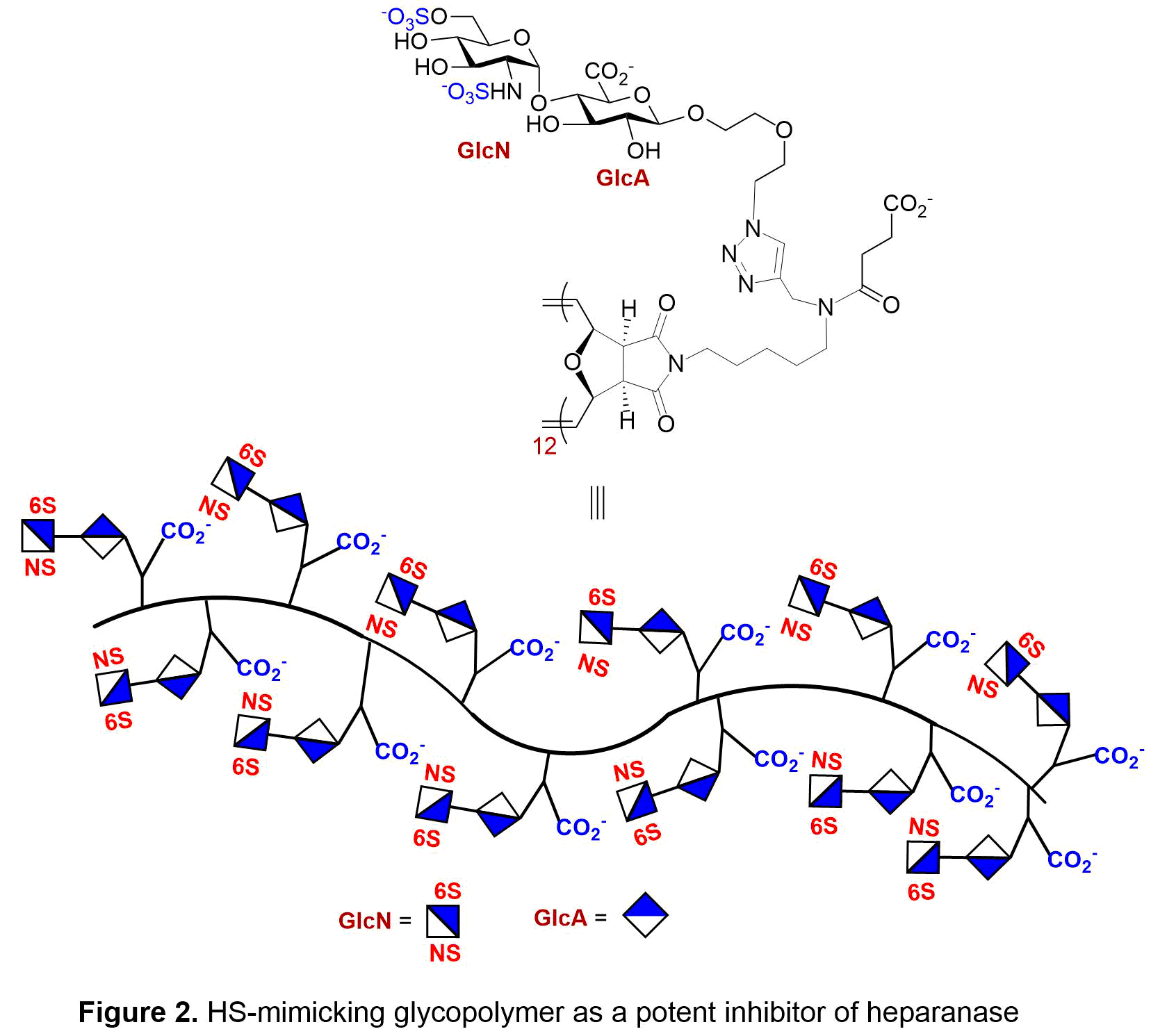
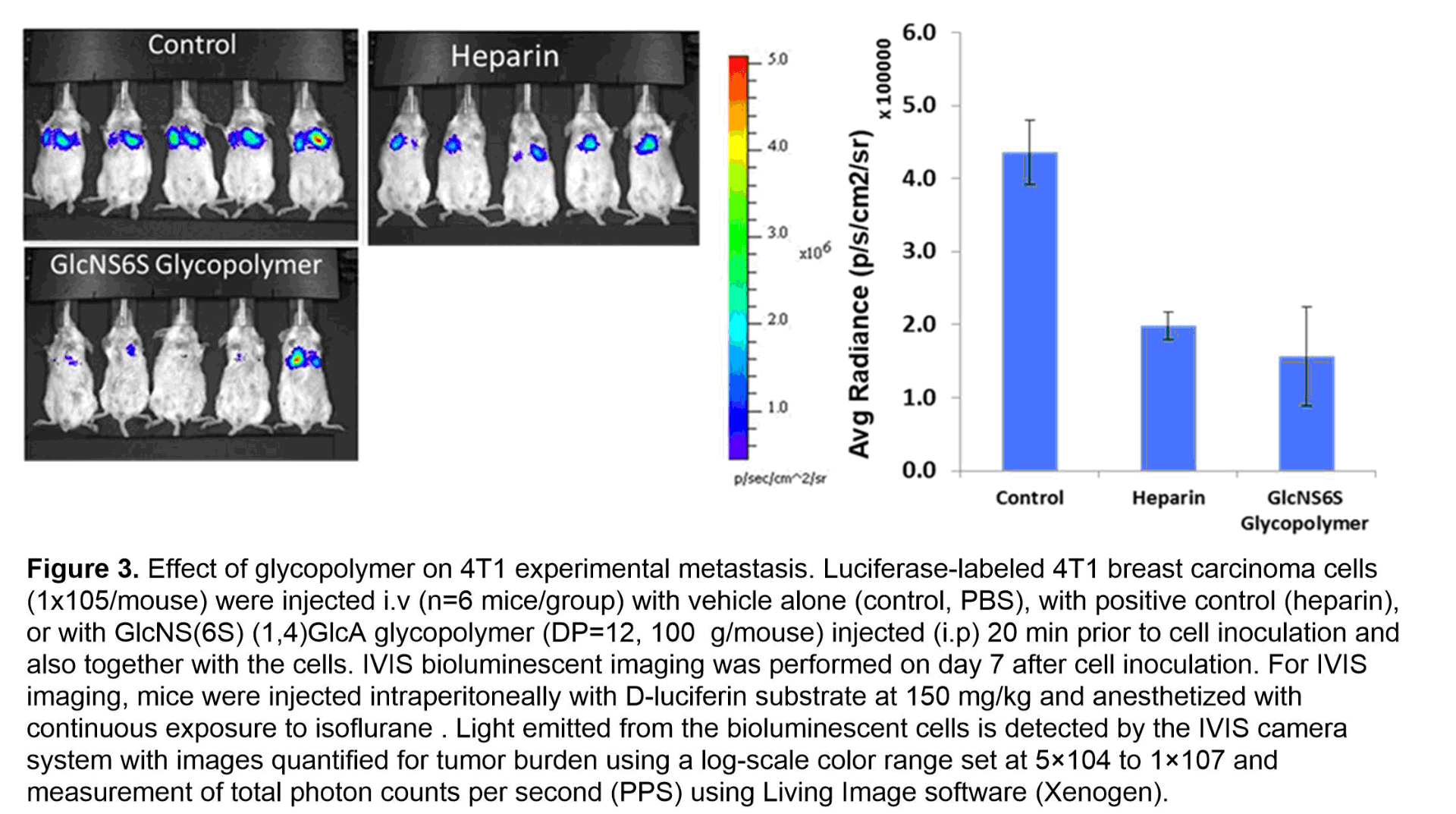
With the desire to inhibit heparanase, we synthesized the sulfated GlcN-GlcA disaccharide, which sits in the -1/-2 subsite of heparanase. We then placed the disaccharide unit onto a multivalent polymerizable scaffold to form glycomonomer. To enhance the overall protein-saccharide avidity and mimic the natural heparan sulfate structure, the synthetic monomer then underwent ring-opening metathesis polymerization (ROMP) to generate the corresponding glycopolymers. The strength of inhibition of heparanase by sulfated 2-aminosugar functionalized polymers were examined in three areas: length of polymer, the display of the saccharides, and the sulfation pattern of the saccharide. From these studies it was determined that the ideal polymer length and saccharide display was a C(6)- and N-sulfated diantennary glycopolymer with a degree of polymerization of 12 repeating units (see Figure 2). In a competitive TR-FRET assay, the GlcNS(6S)a(1,4)GlcA glycopolymer had an IC50 = 0.10 ± 0.04 nM towards heparanase. To ensure heparanase specificity, the most potent glycopolymer inhibitor of heparanase was examined with a solution based competitive biolayer interferometry assay for cross-bioactivity to other HS-binding proteins (growth factors, platelet factor 4, P-selectin) which are responsible for mediating angiogenic activity, antibody-induced thrombocytopenia, and tumor cell metastasis. Compared to heparin, our designed synthetic glycopolymer has a much lower affinity for these proteins. Additionally, the synthetic glycopolymer was shown to have antiproliferative properties when analyzed using a HUVEC cell assay and an anti-metastatic effect in a 4T1 mammary carcinoma model (see Figure 3).
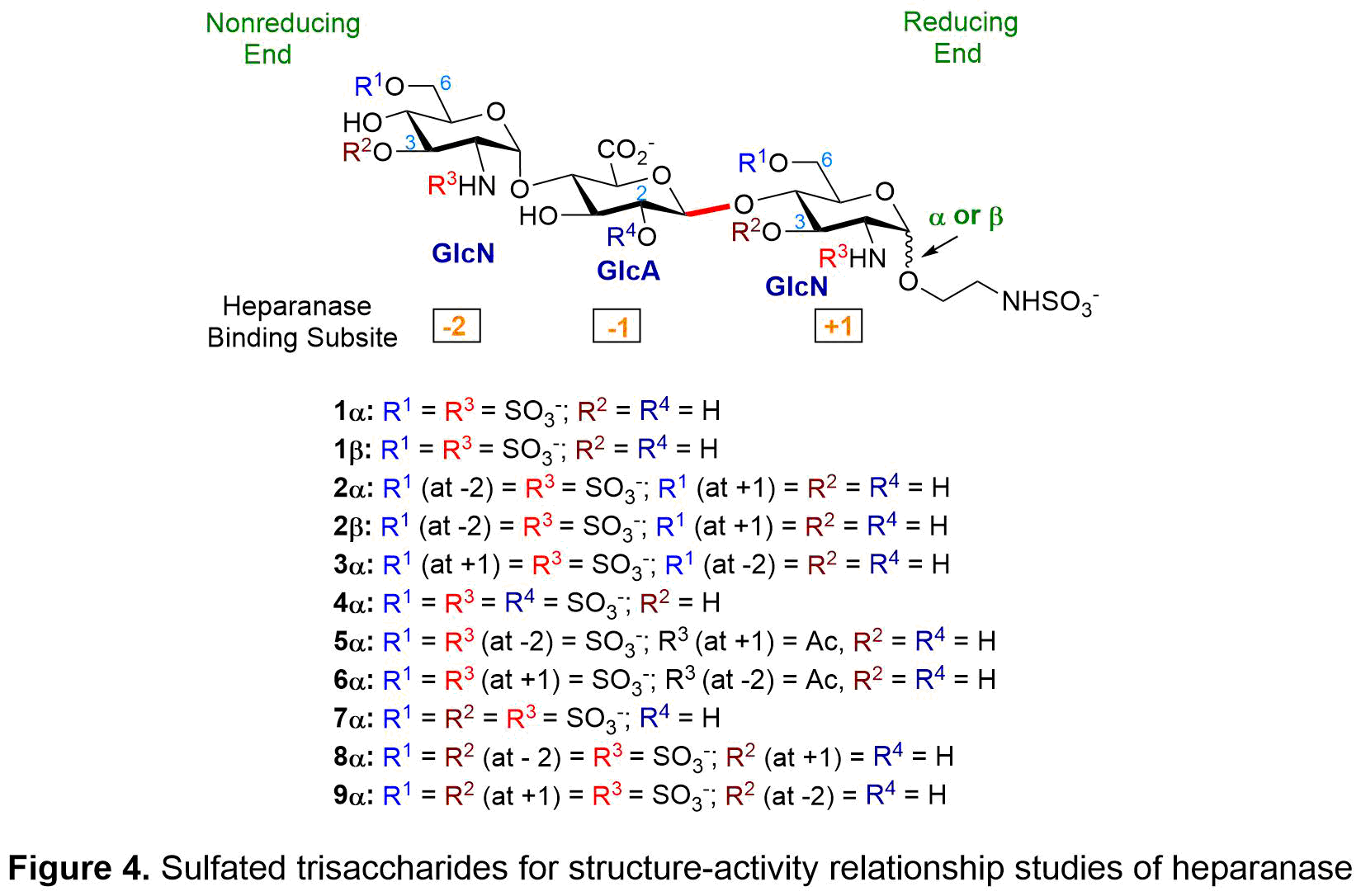
We have synthesized eleven trisaccharides (see Figure 4) that are highly tunable in structure and sulfation pattern, allowing us to determine how heparanase recognizes heparan sulfate (HS) substrate and selects a favorable cleavage site. Our study shows that (1) beta-linked reducing end activated heparanase and was hydrolyzed by heparanase; (2) alpha-linked reducing end inhibited heparanase and was resistant toward hydrolysis; (3) absence of 6-O-SO3– at +1 and at −2 of GlcN (glucosamine) decreased binding to heparanase; (4) N-acetyl groups instead of N-SO3– at the −2 and +1 reduced potency; (5) 3-O-SO3– at +1 and at −2 of GlcN decreased binding to heparanase; and (6) 2-O-SO3– at -1 of GlcA (glucuronic acid) decreased binding to heparanase.
1) Loka, R. S.;** Yu, F.;** Sletten, E. T.;** Nguyen, H. M.* “Design, Synthesis, and Evaluation of Heparan Sulfate Mimicking Glycopolymers for Inhibiting Heparanase Activity.” Chem. Commun. 2017, 53, 9163-9166
2) Sletten, E. T.;** Loka, R. S.;** Yu, F.; Nguyen, H. M.* “Glycosidase Inhibitors from Multivalent Presentation of Heparan Sulfate Saccharides on Bottlebrush Polymer.” Biomacromolecules 2017, 18, 3387-3399
3) Loka, R. S.;** Sletten, E. T.;** Barash, U.; Vlodavsky, I.; Nguyen, H. M.* “Specific Inhibition of Heparanase by Glycopolymer with Well-Defined Sulfation Pattern Prevents Breast Cancer Metastasis in Mice.” ACS Appl. Mater. Interfaces 2019, 11, 244-254
4) Zhu, S.; Li, J.; Loka, R. S.; Vlodavsky, I.; Zhang, K.; Nguyen, H. M. “Modulating Heparanase Activity: Tuning Sulfation Pattern and Glycosidic Linkage of Oligosaccharides” J. Med. Chem. 2020, 63, 4227-4255.
5) Loka, R. S.; Song, Z.; Sletten, E. T. Kayal, Y.; Vlodvasky, I.; Zhang, K.*; Nguyen, H. M.* “Heparan Sulfate-Like Glycopolymer Prevents Pancreatic Beta Cell Destruction and Suppresses Islet Inflammation under the Challenge of Upregulated Heparanase.” ACS Chem. Biol. 2021 (under revision).
SYNTHESIS AND EVALUATION OF CARBOHYDRATE VACCINE ADJUVANTS (Collaborator: Professor Steve Varga – University of Iowa)
The success of vaccination requires the generation of a strong immune response to the inoculated antigens in order to provide long-term protective immunity against infectious diseases. To achieve this goal often requires the use of an adjuvant, a substance that boosts the body’s immune response to the vaccine. Unfortunately, there are only a few human vaccine adjuvants with an extensive safety record and minimal toxicity approved for clinical use. Therefore, there is a great demand to develop novel adjuvants that not only significantly enhances the immune response to a co-administered antigen, but also must be minimally toxic for clinical use, cost-effective, and stable for long-term storage.
In efforts to discover novel vaccine adjuvants, an in vivo screening of 47 saponins from medicinal plants for their immunostimulatory and hemolytic activities has led to the discovery of new exciting vaccine adjuvants. Among 47 saponin natural products evaluated, soyasaponins have emerged as the most potent adjuvants. These newly-discovered saponins exhibited a remarkable adjuvant activity with almost negligible toxicity. However, obtaining soyasaponins from natural sources is a complicated process of extraction and HPLC purification that result in the production of minute. As such, isolation of soyasaponins is economically unfeasible and unsustainable if sufficient quantities are required for immunological evaluation and clinical applications. Since the FDA has strict regulations regarding to the purity and quality of vaccine adjuvants for use in humans, a synthetic source must be developed for soyasaponins to be utilized as clinically relevant adjuvants. The goal of our project is to direct the chemical synthesis and preclinical evaluation of soyasaponins (see Figure 5), which hold promise as potent adjuvants with negligible toxicity for vaccine therapeutics.

COUNTERING TAUOPATHY WITH HEPARAN SULFATE DERIVATIVES (Collaborators: Prof. Min-Hao Kuo at Michigan State University and Kezhong Zhang at Wayne State University)
Six million people in the US are suffering from Alzheimer’s disease (AD). On average, each patient is under the care of three unpaid caregivers, resulting in billions of dollars of additional economic burden. Tremendous efforts have been devoted to dissecting the disease mechanisms. Despite the recurring failures in clinical trials, it is hopeful that better understanding of key pathological players and advancement of technology in diagnosis and drug discovery will lead to successes in fighting this disease.
AD is one of 20 neurodegenerative tauopathies that share the conspicuous fibrillar hyperphosphorylated tau (p-tau) deposits in the brain. The spatiotemporal distribution of tau inclusions correlates with the advancement of neurological manifestations. A familial form of frontotemporal lobar dementia is caused by mutations in the MAPT gene for tau protein, lending the strongest support for a causal role of tau mis-regulation. Consistently, intracerebral injection of abnormally phosphorylated tau (p-tau) isolated from patients or transgenic mice recapitulated the pathology in animal brains. Accumulating evidence suggests that the pre-tangle, oligomeric p-tau causes AD pathology in a diffusible, prion-like fashion. Firstly, a fly model showed transgenic tau-dependent pathology without the formation of tangles. Diffusion of the pathogenic p-tau species may be through a vesical-free mechanism, or by exocytosis. Also, p-tau may attack cells by multiple mechanisms as well. For example, a pore-like structure of tau annular protofibrils was found on the membrane of human tauopathy brain samples. Bulk endocytosis of oligomeric p-tau by neurons was reported. The hypothesis of soluble cytotoxic aggregation intermediates is gaining momentum.
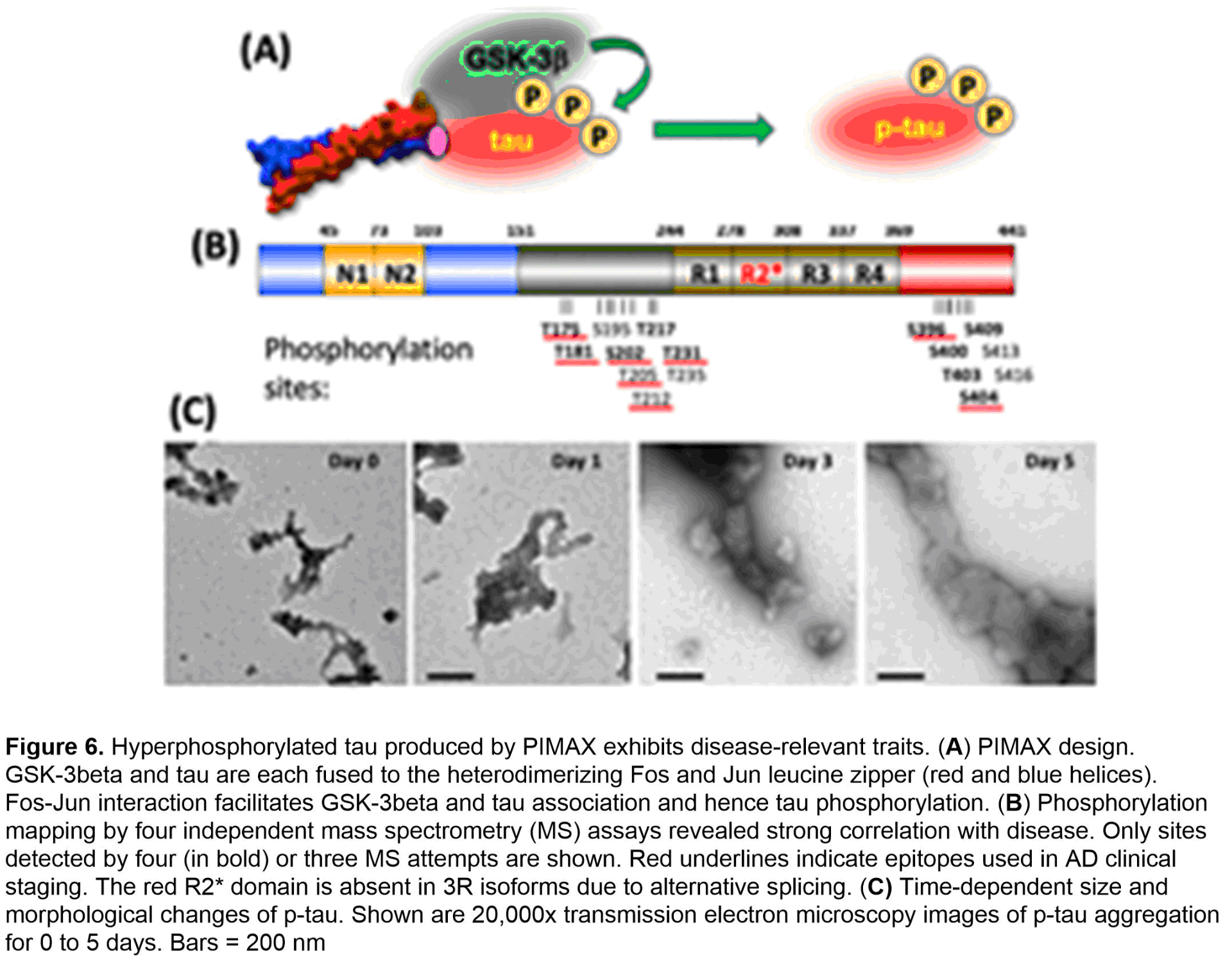
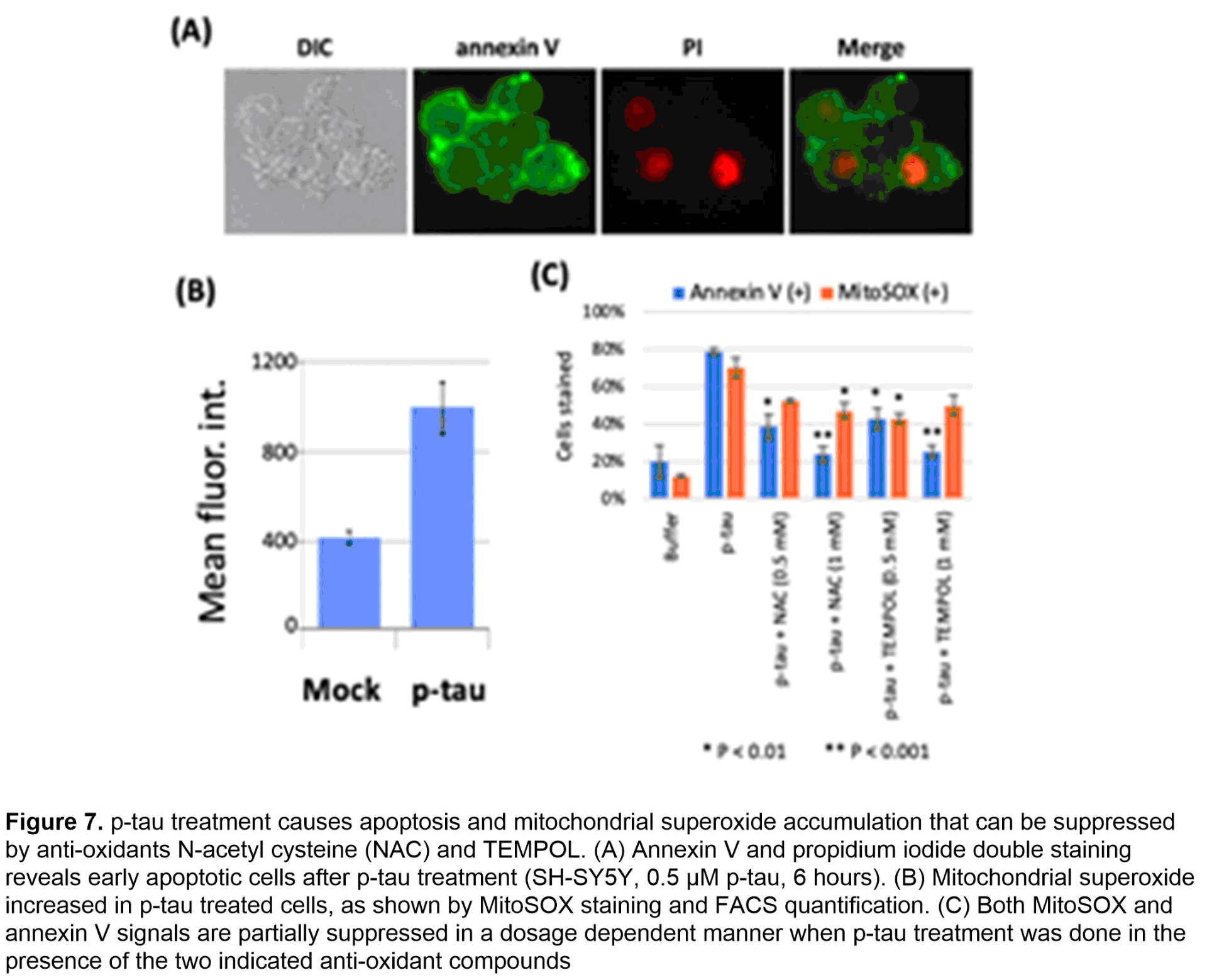
To procure a pathophysiologically relevant p-tau protein of high quality and quantity that supports AD mechanistic and drug discovery research, we have developed and used the PIMAX system (Figure 6A). The resultant p-tau was examined by a variety of assays. Mass spectrometry confirms the disease relevance in that of the 16 sites detected by at least 3 of 4 independent mass spectrometry tests, 8 are AD staging markers (red underlines, Figure 6B). Biochemically, p-tau spontaneously organizes from fractals to much larger and ordered structures in 5 days (Figure 6C). Critically, the aggregation intermediates of p-tau cause cell death at sub-micromolar concentrations by triggering apoptosis (Figure 7A) and the accumulation of mitochondrial superoxide (Figure 7B). Co-treating cells with selective anti-oxidants alleviates superoxide accumulation and apoptosis (Figure 7C), suggesting a path of destruction as the following: p-tau uptake → mitochondrial dysfunction → superoxide accumulation → apoptosis. Besides apoptosis and mitochondrial dysfunction, additional cellular defects have been linked to AD neurodegeneration, most notably, the endoplasmic reticulum (ER) stress, unfolded protein responses, and inflammation.
CATALYTIC STEREOSELECTIVE GLYCOSYLATIONS
Carbohydrates are widespread in nature and have been considered as the frontier of medicinal chemistry. In general, the sugar based biomolecules are constructed from rudimentary glycosylation reactions, which take place between a glycosyl donor (electrophile) and glycosyl acceptor (nucleophile). These reactions allow for the establishment of two different a- and b-stereoisomers that differ in the configuration of the anomeric carbon. In many cases, alpha-glycosides have a cis relationship between the substituents on the anomeric carbon and the second carbon of the electrophilic coupling partner; with the exclusion of a few rare sugars. Conversely, beta-glycosides would have a 1,2 trans relationship with the same exceptions. By taking advantage of neighboring group participation of acyl protecting groups at the second carbon of glycosyl donor, beta-1,2-trans glycosides can be made with high levels of selectivity. For this reason, many methods focuses on the development of the stereoselective formation of alpha-1,2-cis linked glycosides, which are important components of bioactive oligosaccharides. The stereoselective synthesis of an alpha-1,2-cis glycoside, however, has proven to be a hurdle when utilizing traditional modes of carbohydrate activation. There have been many strategies developed to address this issue. Unfortunately, current methods often rely on the nature of the protecting groups bound to the electrophile to influence selectivity, thereby making them highly specific for each electrophilic coupling partner. On the other hand, catalytic approaches to alpha-1,2-cis glycosidic bond are relatively limited, although they could provide catalyst control without relying on the influence of the protecting group nature of carbohydrate substrates.
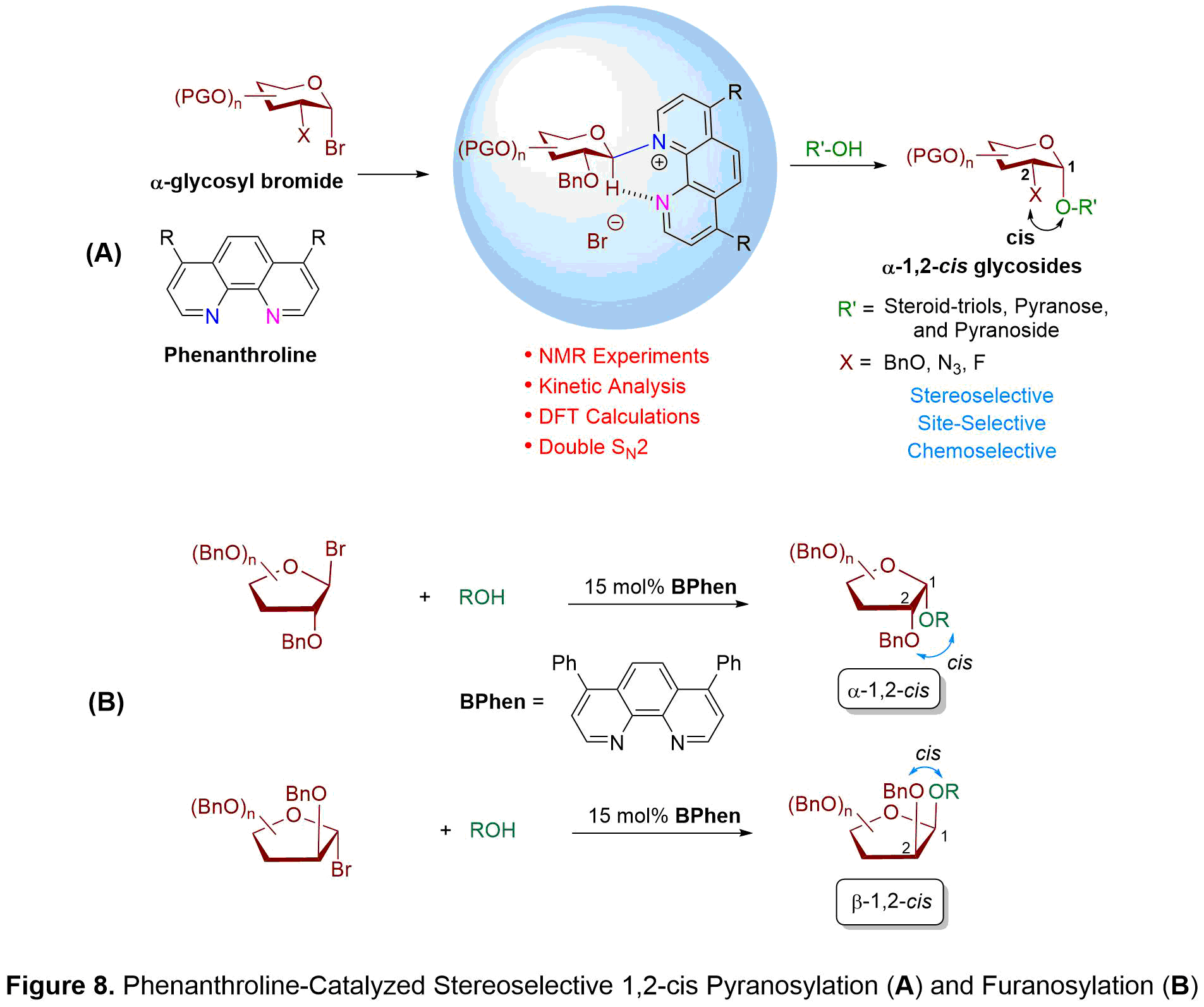
We discovered that phenanthroline, a rigid and planar compound with two fused pyridine rings, could be used as a nucleophilic catalyst to efficiently access high yielding and diastereoselective alpha-1,2-cis glycosides through the coupling of hydroxyl acceptors with alpha-glycosyl bromide donors (see Figure 8A). We have conducted an extensive investigation into the reaction mechanism, wherein the two glycosyl phenanthrolinium ion intermediates, a 4C1 chair-liked beta-conformer and a B2,5 boat-like alpha-conformer, have been detected in a ratio of 2:1 (beta:alpha) using variable temperature NMR experiments. Furthermore, NMR studies illustrate that a hydrogen bonding is formed between the second nitrogen atom of phenanthroline and the C1-anomeric hydrogen of sugar moiety to stabilize the phenanthrolinium ion intermediates. To obtain high alpha-1,2-cis stereoselectivity, a Curtin-Hammett scenario was proposed wherein interconversion of the 4C1 chair-like beta-conformer and B2,5 boat-like alpha-conformer is more rapid than nucleophilic addition. Hydroxyl attack takes place from the alpha-face of the more reactive 4C1 beta-phenanthrolinium intermediate to give an alpha-anomeric product. The phenanthroline catalyst is also effective at promoting stereoselective 1,2-cis furanosylations (see Figure 8B). NMR experiments and density-functional theory calculations support an associative mechanism in which the rate-determining step occurs from an invertive displacement of the faster reacting phenanthrolinium ion intermediate with alcohol nucleophile. The phenanthroline catalysis system is applicable to a number of furanosyl bromide donors to provide the challenging 1,2-cis substitution products in good yield with high anomeric selectivities. While arabinofuranosyl bromide provides beta-1,2-cis products, xylo- and ribofuranosyl bromides favor alpha-1,2-cis products.
1) Yu, F.; Li, J.; DeMent, P. M.; Tu, Y.-J.; Schlegel, H. B.; Nguyen, H. M. “Phenanthroline-Catalyzed Stereoselective Glycosylations.” Angew. Chem. Int. Ed. 2019, 58, 6957-6961.
2) Yu, F; Dickson, J. L.; ** Loka, R. S.; ** Xu, H; ** Schaugaard, R. N.; Schlegel, H. B; Luo, L;* Nguyen, H. M.,* “Diastereoselective sp3 C-O Bond Formation via Visible Light-Induced, Copper-Catalyzed Cross Couplings of Anomeric Alkyl Bromides with Aliphatic Alcohols.” ACS Catalysis 2020, 10, 5990-6001.
3) DeMent, P. M.; Wakpal, J.; Lu, C.; Schaugaard, R. N.; Schlegel, H. B.; Nguyen, H. M. “Phenanthroline-Catalyzed Stereoselective Formation of alpha-1,2-cis-2-Deoxy-2-Fluoro Glycosides.” ACS Catalysis 2021, 11, 2108-2120.
4) Li, J.; Nguyen, H. M. “A Mechanistic Probe into 1,2-cis Glycoside Formation Catalyzed by Phenanthroline and Further Expansion of Scope” Adv. Synth. Catal. 2021, 363, 4054-4066.
5) Xu, H.; Schaugaard, R. N.;** Li, J.;** Schlegel, H. B.:* Nguyen, H. M.* “Phenanthroline-Catalyzed Stereoselective 1,2-cis Furanosylations.” J. Am. Chem. Soc. 2021 (under revision).
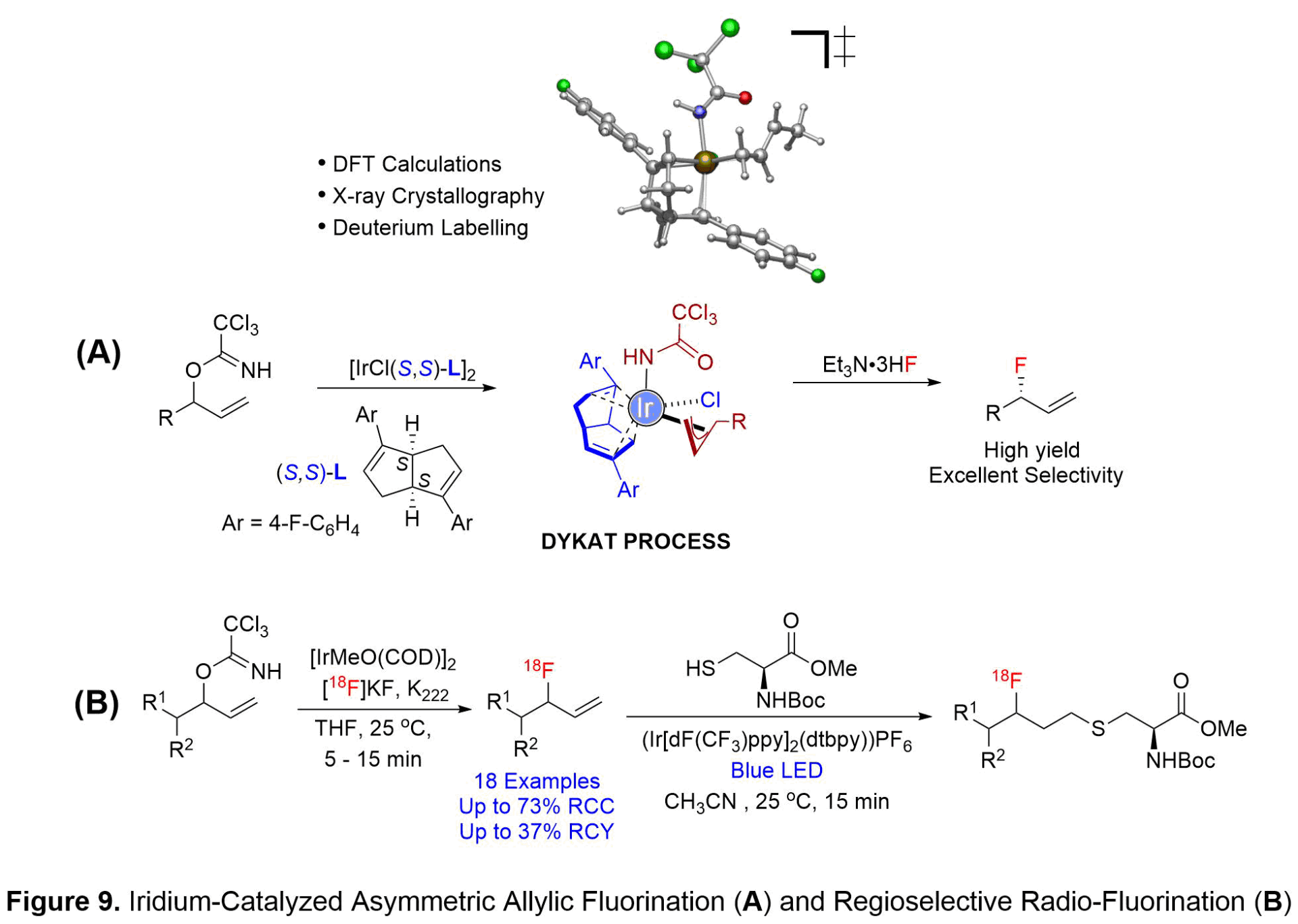
IRIDIUM CATALYZED ASYMMETRIC ALLYLIC FLUORINATION AND REGIOSELECTIVE ALLYLIC RADIO-FLUORINATION (Collaborator: Prof. David Dick at University of Iowa PET Imaging Center)
The incorporation of fluorine atoms has far-reaching impacts in organic chemistry. Over the past decade, molecules containing carbon-fluorine bonds have become increasingly prevalent in pharmaceutical, agricultural, and materials chemistry. Currently, 20% of pharmaceutical targets and 30% of agrochemicals on the market contain at least one carbon-fluorine bond. The introduction of a fluorine atom into biologically active molecules has the potential to improve a number of factors including absorption, metabolism, and potency of the drug candidate. A significant amount of research has been conducted on the selective incorporation of fluorine into a variety of functional groups. However, the formation of allylic fluorides has been significantly understudied, leading to a need for the creation of new methodologies. Transition metal catalysis has come to the forefront as a viable option to selectively produce these carbon-fluorine bonds.
Allylic fluorides are key scaffolds in a variety of biologically relevant molecules. Traditional methods to form allylic fluorides lack the ability to produce a single regio-isomer, and instead lead to a mixture of linear and branched products. The creation of one single enantiomer is of paramount importance as different enantiomers can have variable affects in vivo. We discovered racemic allylic trichloroacetimidates as competent electrophiles in a chiral bicyclo[3.3.0]octadiene-ligated iridium-catalyzed asymmetric fluorination with Et3N·3HF (see Figure 9A). The methodology represents an effective route to prepare a wide variety of alpha-linear, alpha-branching, and beta-heteroatom substituted allylic fluorides in good yields, excellent branched-to-linear ratios, and high levels of enantioselectivity. Additionally, the catalytic system is amendable to the fluorination of optically active allylic trichloroacetimidate substrates to afford the fluorinated products in good yields with exclusively branched selectivity. Excellent levels of catalyst-controlled diastereoselectivities using either (R,R) or (S,S)-bicyclo[3.3.0]octadiene ligand are observed. The synthetic utility of the fluorination process is illustrated in the asymmetric synthesis of 15-fluorinated prostaglandin and neuroprotective agent P7C3-A20.
The iridium-catalyzed allylic fluorination reaction mechanism was investigated by conducting a synergistic computational and experimental study in collaboration with the Gutierrez group (University of Maryland). Mechanistic studies revealed the critical role of the trichloroacetimidate acting as both a leaving group and a ligand. Initial ionization of the allylic trichloroacetimidate occurs with retention of configuration due to the pre-coordination of the iridium metal to the trichloroacetimidate substrate (see Figure 9A). The ligated trichloroacetimidate-iridium complex can undergo equilibration between the two diastereomeric pi-allyl-iridium complexes through a dynamic kinetic asymmetric transformation (DYKAT)-like mechanism allowing for formation of an enantioenriched allylic fluoride in high regioselectivity and asymmetric induction. Deuterium scrambling experiments were utilized to determine that fluoride addition occurs via an outer-sphere nucleophilic addition.
We further translated our methodology for the synthesis of allylic [18F]fluorides that could be used for positron emission tomography (PET) imaging. PET imaging has become a powerful tool for the qualitative and quantitative assessment of metabolically active diseases. Specifically, these PET radiotracers are used for the imaging of a variety of cancers, and cardiovascular and neurological disorders. We first investigated a variety of transition metal catalysts, including both rhodium and iridium species. Iridium was found to be the optimal metal catalyst, providing the desired substrate in moderate radiochemical conversion (see Figure 9B). The optimized reaction conditions were implemented for a variety of substrates, resulting in the desired radio-labeled products in good radiochemical yields and in short reaction times (5-15 minutes). Several substrates were isolated to determine the radiochemical yield and the molar activity (ratio of radiolabeled product to non-radiolabeled product). To show the practicality of the method, thiol-ene “click” chemistry was used to couple an amino acid to a radio-labeled substrate (see Figure 9B). Our studies are significant as this methodology has the potential to lead to new radio-tracers for the labeling of peptides and amino acids for PET imaging.
1) Topczewski, J. J.; Tewson, T. J.; Nguyen, H. M. “Iridium-Catalyzed Allylic Fluorination of Trichloroacetimidates.” J. Am. Chem. Soc. 2011, 133, 19318-19321.
2) Zhang, Q.; Nguyen, H. M. “Rhodium-Catalyzed Regioselective Ring Opening of Vinyl Epoxides with Et3N.3HF: Formation of Allylic Fluorohydrins.” Chem. Sci. 2014, 5, 291-296.
3) Zhang, Q; Stockdale, D. P.; Mixdorf, J. C.; Topczewski, J. J.; Nguyen, H. M. “Iridium-Catalyzed Enantioselective Fluorination of Racemic, Secondary Trichloroacetimidates.” J. Am. Chem. Soc. 2015, 137, 11912-11915.
5) Mixdorf, J. M.; Sorlin, A. M.; Nguyen, H. M. “Asymmetric Synthesis of Allylic Fluorides via Fluorination of Racemic Allylic Trichloroacetimidates Catalyzed by a Chiral Diene-Iridium Complex.” ACS Catal., 2018, 8, 790-801.
6) Mixdorf, J. C;** Sorlin, A. M.;** Dick, D.;* Nguyen, H. M.* “Iridium-Catalyzed Radiosynthesis of Branched Allylic [18F]Fluorides.” Org. Lett. 2019, 21, 60-64.
7) Sorlin, A. M.;** Mixdorf, J. C.;** Rotella, M.; Martin, R.; Gutierrez, O.;* Nguyen, H. M.* “The Role of Trichloroacetimidate to Enable Irididium-Catalyzed Regio- and Enantioselective Allylic Fluorination.” J. Am. Chem. Soc. 2019, 143, 14843-14882.
8) Sorlin, A. M.; Fuad, U. O.; English, C. K.; Nguyen, H. M.* “Advances in Nucleophilic Allylic Fluorination.” ACS Catal. 2020, 10, 11980-12010.
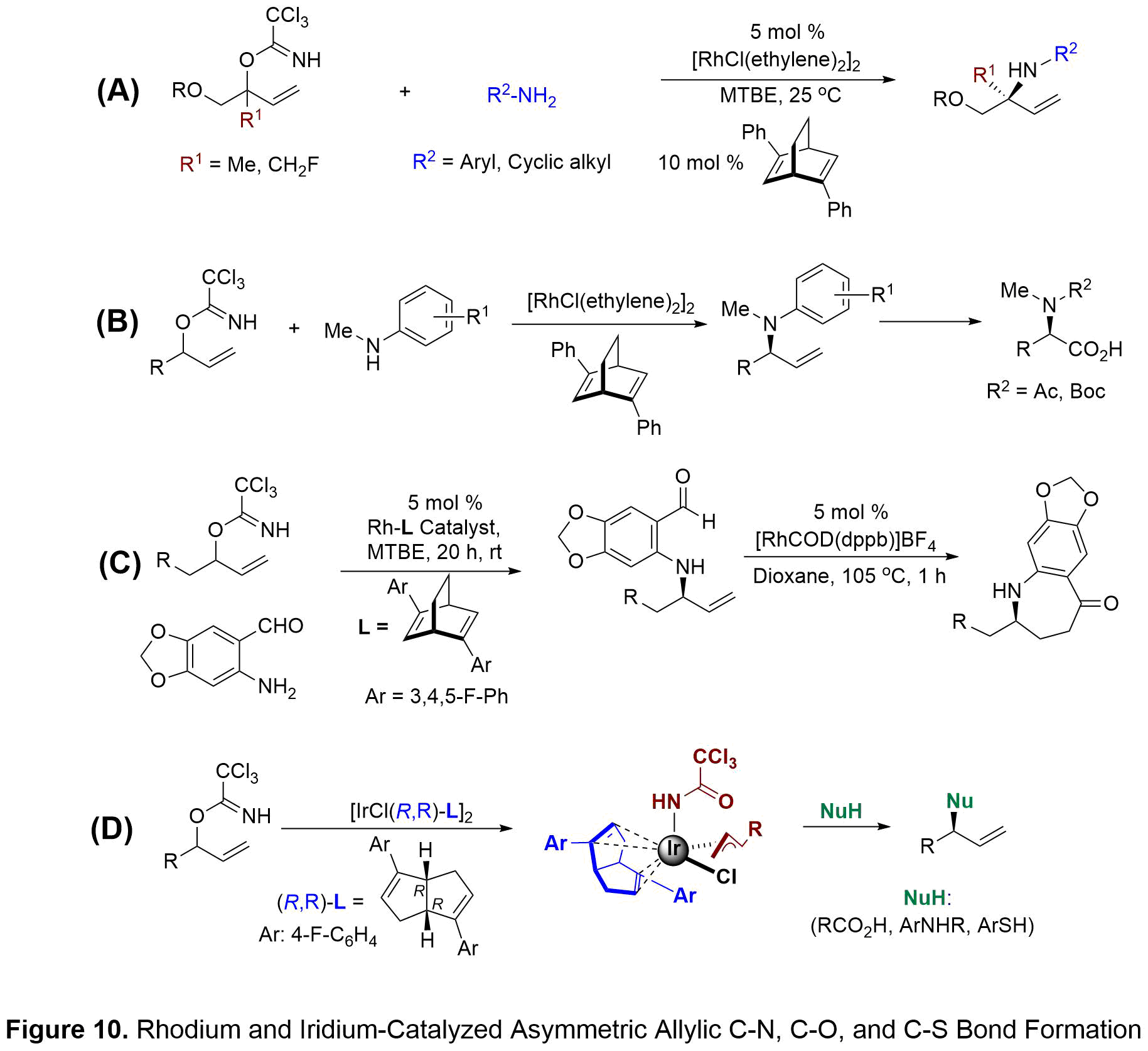
RHODIUM/IRIDIUM CATALYZED ASYMMETRIC ALLYLIC C-N, C-O, AND C-S BOND FORMATION
Transition-metal-catalyzed allylic substitutions have become a useful tool for enantioselective construction of chiral C-O, C-N, and C-S motifs that are found in a wide variety of natural products, pharmaceuticals, and agrochemicals. Our group has focused on the development of a NEW method that ultimately resulted in the first reported dynamic kinetic asymmetric transformation (DYKAT) of acyclic tertiary allylic electrophiles with aniline and cyclic amine nucleophiles (see Figure 10A). Hayashi’s diphenyl bicyclo[2.2.2]octadiene was the most efficient ligand in the DYKAT reaction with highest yields and asymmetric induction. This methodology allows for the high-yielding, regio- and enantioselective synthesis of quaternary amine-containing centers. This operationally simple process is catalyzed by a chiral diene-ligated rhodium(I) complex and utilizes easily prepared branched trichloro-acetimidates and unactivated anilines. The synthetic utility of this method was then illustrated by application to the preparation of both enantioenriched amino acid derivatives and nitrogen-containing heterocycles.
Secondary allylic trichloroacetimidates work equally well in the DYKAT reaction for preparation of tertiary carbon centers containing an amine functionality. My studies focused primarily on secondary acyclic anilines which are known to provide the amination products with low regioselectivity in previous asymmetric methods.1 Under our DYKAT conditions, Hayashi’s diphenyl bicyclo[2.2.2]octadiene ligand provided N-methyl arylamine products in high yields and selectivities (see Figure 10B). The utility of this methodology was demonstrated by preparing N-methyl homophenylalanine derivatives found in antillatoxin B. Utilization of primary anilines and recently N-methyl benzylamine with secondary allylic trichloroacetimidates shows the generality of the DYKAT reaction for the selective synthesis of enantiopure allylic amines. Our methodology to the enantioselective formation of 7-membered aza-ketones (see Figure 10C). This methodology relies on sequential DYKAT amination using 2-amino benzaldehyde nucleophiles followed by rhodium-catalyzed hydroacylation of the enantioenriched allylic aryl amines. Our approach to these medium-sized rings differs from prior reports because asymmetric induction is substrate-controlled versus the use of chiral catalysts in the hydroacylation step. Following optimization of the sequential amination and hydroacylation steps, a number of enantiopure dihydro-benzoazepin-5-ones were obtained in 50-94% overall yield with excellent enantioselectivites. Importantly, complete transfer of chirality from amination to hydroacylation products was observed in all cases.
Recently, our group showed that in the presence of a chiral bicyclo[3.3.0]octadiene-ligated Ir catalyst and Et3N•3HF as a nucleophilic fluoride, racemic branched allylic trichloroacetimidates undergo rapid asymmetric fluorination. Our studies on the chiral diene-ligated iridium catalyst in the fluorination lead us to question whether this iridium-trichloroacetimidate system could be capable of constructing C-O, C-N, and C-S bonds in an enantioselective fashion. Herein, we report a new strategy involving the chiral diene-ligated Ir-catalyzed asymmetric substitutions of racemic, branched allylic trichloroacetimidates with a wide range of heteroatom nucleophiles (see Figure 10D). These nucleophiles include aromatic carboxylic acids, alkyl carboxylic acids, aromatic amines, and aromatic thiols. Reactions of these heteroatom nucleophiles lead to the highly branched-selective allylic substitution products in good yields and high levels of enantioselectivities.
1) Arnold, J. S.; Nguyen, H. M.* “Rhodium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Racemic Tertiary Allylic Trichloroacetimidates with Aniline Nucleophiles.” J. Am. Chem. Soc. 2012, 134, 8380 – 8383.
2) Arnold, J. S.; Cizio, G. T.;*** Heitz, D. R.;*** Nguyen, H. M.* “Rhodium-Catalyzed Enantioselective Amination of Racemic Secondary Allylic Trichloroacetimidates with N-Methyl Anilines.” Chem. Commun. 2012, 48, 11531 – 11533.
3) Arnold, J. S.;** Mwenda, E.;** Nguyen, H. M.* “Sequential Amination and Hydroacylation Reactions for the Enantioselective Synthesis of Seven-Membered Heterocycles.” Angew. Chem. Int. Ed. 2014, 53, 3688 – 3692.
4) Mwenda, E.; Nguyen, H. M.* “Enantioselective Synthesis of 1,2-Diamines via Rhodium-Catalyzed Enantioselective Dynamic Asymmetric Transformations of Racemic Allylic Trichloroacetimidates.” Org. Lett. 2017, 19, 4814 – 4817.
5) Arachchi, M. K.; Nguyen, H. M.* “Iridium-Catalyzed Enantioselective Allylic Substitutions of Racemic, Branched Trichloroacetimidates with Heteroatom Nucleophiles: Formation of Allylic C-O, C-N, and C-S Bonds” Adv. Synth. Catal. 2021, 363, 4239-4246.