Research at a glance:
As a fundamental approach to our research, we develop hypotheses utilizing the powerful genetic tools of the yeast system, then further test/validate our findings using relevant mammalian cell lines. Our research can be broadly divided into two major projects: Barth syndrome & cardiolipin and bipolar disorder & inositol. Below is an overview of each project, followed by additional details and current funding sources.
Barth Syndrome & Cardiolipin
In the United States, 1,000 to 4,000 children are born each year with a chronic mitochondrial disease, and at the global level, mitochondrial diseases have an estimated prevalence of one in every 5,000 individuals (~1.6 million cases worldwide). Furthermore, these numbers do not take into account the many other disorders that are associated with metabolic alterations and secondary mitochondrial dysfunction. Thus, a prerequisite to understanding the pathological features of, and developing treatments for, many chronic diseases is to gain an understanding of the specific mitochondrial/metabolic pathways that are dysregulated in the disease state.
As the hallmark phospholipid of mitochondria, cardiolipin (CL) is vitally important for both mitochondrial function and overall cellular homeostasis. This is perhaps best illustrated by the rare genetic disorder Barth syndrome (BTHS), which is caused by mutations in the CL remodeling enzyme, tafazzin. BTHS is characterized clinically by a combination of heart defects (cardiomyopathy), diminished muscle tone (hypotonia/skeletal myopathy), and reduced white blood cell count/immune function (neutropenia). The TAFAZZIN gene is located on the X chromosome, and because males only possess one copy of the X chromosome, BTHS almost exclusively affects men.
Although the general importance and functions of CL have been well-studied, a critical gap in our understanding of BTHS is how the specific molecular changes in CL (resulting from TAFAZZIN mutations) translate into the clinical pathologies of the disease (cardiomyopathy, skeletal myopathy, and neutropenia). Our research aims to address this knowledge gap by studying the cellular consequences of CL/tafazzin deficiency using various established models of BTHS, including yeast, mouse, and human cell lines, as well as mouse tissue. We anticipate that these studies will identify biological pathways that can serve as new therapeutic targets for treating BTHS (and potentially other mitochondrial/metabolic diseases).
See below for more details on this project.
Bipolar Disorder & Inositol
Collectively, chronic neurological disorders have been estimated to affect over one billion people worldwide (roughly one in six individuals). Due to the complexity of the central nervous system, much remains unknown regarding the pathophysiology of most neurological disorders. This is highlighted by the fact that many of the most widely prescribed drugs to treat such diseases have unknown mechanisms of action and vary substantially in their efficacy from one patient to another.
A prime example of this is observed in bipolar disorder (BD), which affects approximately 1-2% of people worldwide. The exact cause of BD is unknown. Historically, the most popular drugs used to treat BD include the mood stabilizers valproic acid/valproate (VPA), lithium (Li), and carbamazepine. Interestingly, the ability of these drugs to treat BD was discovered fortuitously (i.e., they were not purposefully “designed” to treat the disorder), and surprisingly, despite decades of research, the mechanism(s) by which they operate remain unknown. Unfortunately, these drugs are not effective for many patients and can cause severe, life-threatening side effects in others, underscoring the need for new therapeutic approaches to manage BD. Gaining an understanding of how current BD drugs mediate their therapeutic effects will offer clues that can be used in the development of better drugs, and to this end, it is intriguing that a common cellular effect of the above mood stabilizers is a reduction in levels of the simple sugar, inositol.
In light of this, our lab studies the cellular effects of both inositol deprivation and mood stabilizer treatment using yeast, mouse, rat, and human cell lines. We are also interested in understanding how cells regulate inositol synthesis. We have recently developed a new set of biological “tools” to facilitate this research, including a human cell line that cannot synthesize inositol, an inositol-free cell culture system, and a yeast-based bioassay for measuring the concentration of inositol in unknown samples. We anticipate that this research will help to identify the mechanism(s) of action for currently available mood stabilizers and guide the development of new drugs for treating bipolar disorder that exhibit greater efficacy and less side effects. Furthermore, as altered levels of inositol are associated with many other human diseases, our studies investigating the regulation of inositol synthesis will be of broad interest translationally.
See below for more details on this project.
Join us!
Individuals with an interest in joining our research team or learning more about the techniques we utilize in the lab should check out our prospective students page. For additional information about our specific ongoing research projects, please contact our current students or Dr. Greenberg.
Ongoing research projects and funding:
Elucidating the role of cardiolipin in mitochondrial and cellular functions
Cardiolipin (CL), the signature phospholipid of mitochondria, is crucial not only for mitochondrial function but also for a plethora of cellular processes that are not associated with mitochondrial bioenergetics. The de novo synthesis of CL is followed by a remodeling cycle of deacylation (removal of a fatty acid) followed by reacylation. Specific fatty acyl composition is acquired during this process, and remodeled CL contains predominantly unsaturated fatty acids. The importance of CL remodeling is underscored by the life-threatening genetic disorder Barth syndrome (BTHS), caused by mutations in tafazzin, the enzyme that reacylates CL.
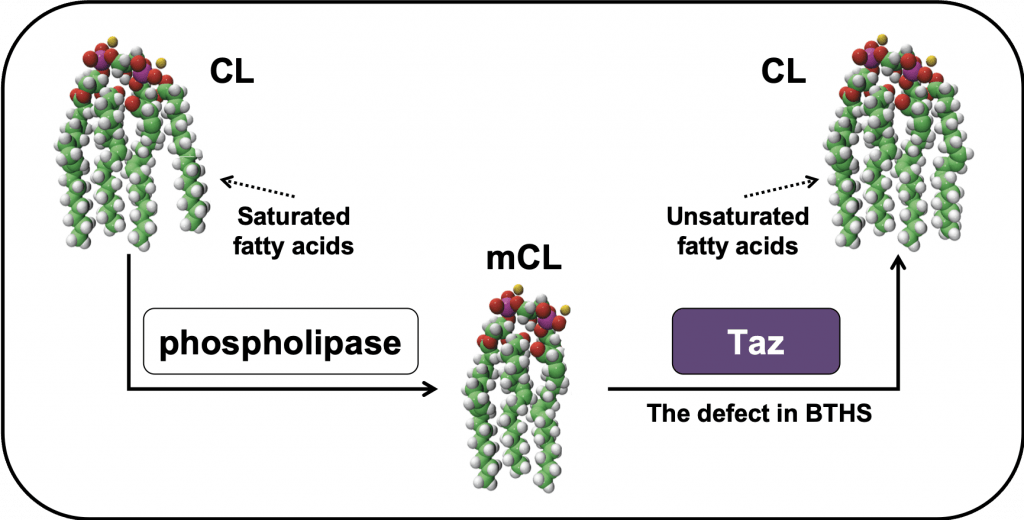
CL-deficient mutants have been instrumental in elucidating the functions of this unique lipid. Using yeast, we identified the first CL mutant (crd1Δ), which cannot synthesize CL. We also developed the yeast model for BTHS, the taz1Δ mutant, and a mouse myoblast BTHS model, TAZ-KO, both of which lack tafazzin. Utilizing these mutants, we have determined that CL homeostasis regulates mitochondrial bioenergetics; mitochondrial lipid peroxidation and reactive oxygen species generation; intermediary metabolism; iron-sulfur biogenesis; and myoblast differentiation/myogenesis.
Our current studies are focused on understanding how aberrant CL remodeling (at the molecular level) translates into the diverse clinical pathologies associated with BTHS (at the organ system/organismal level). This knowledge will provide guidance for the development of targeted therapies to more effectively treat BTHS patients. Our approach is to utilize the power of the yeast model to develop hypotheses that can be tested in appropriate mammalian systems. Specifically, our research addresses the following fundamental questions:
- How does tafazzin/CL deficiency lead to the various BTHS pathologies?
- What is the evolutionary fitness advantage (i.e., “purpose”) of CL remodeling?
- What physiological modifiers affect the severity of BTHS?
Current funding for this project:
NIH – R01 GM134715 – Controlling Monolysocardiolipin/Cytochrome c Peroxidase Complexes in Barth Syndrome
NIH – R01 HL117880 – The Role of Cardiolipin in the TCA Cycle: Implications for Barth Syndrome
Cayman Biomedical Research Institute – Evaluation of irisin as a potential treatment for impaired myogenesis in Barth syndrome
Cellular consequences of inositol depletion and mood stabilizer treatment
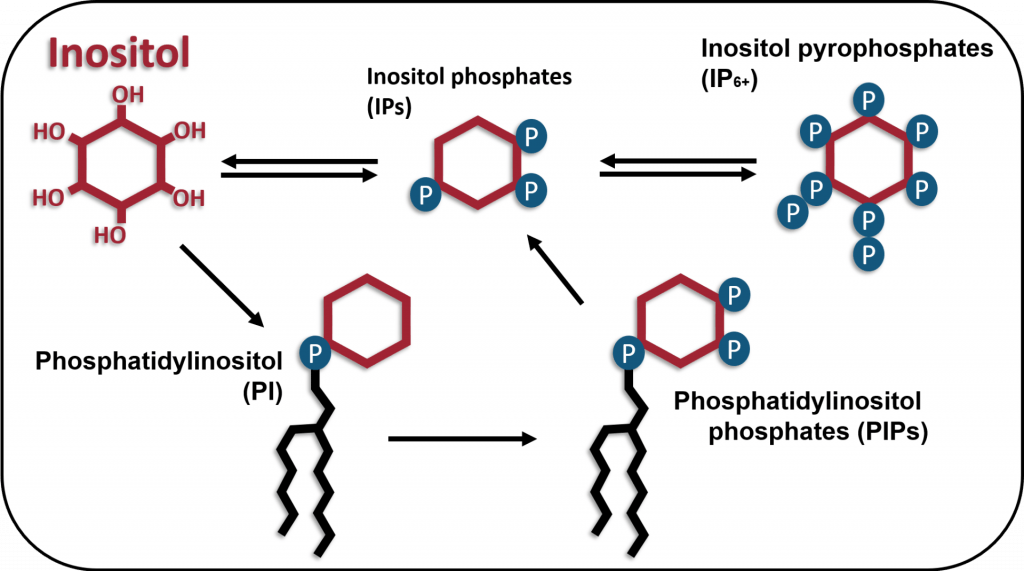
Inositol is an essential metabolite that plays a fundamental role in regulating cellular processes. The phosphorylation of inositol generates numerous inositol phosphates and phosphoinositides, many of which are signaling molecules that control membrane trafficking, calcium mobilization, chemotaxis, ion channel activity, cytoskeletal organization, and gene expression. Furthermore, phosphatidylinositol is a precursor for the synthesis of sphingolipids, which play an important role in signal transduction and trafficking. Consistent with its essential role in cellular function, inositol deprivation leads to a rapid loss of viability. Therefore, cells must obtain inositol either from extracellular sources or through de novo synthesis, mediated in part by the rate-limiting enzyme myo-inositol-3-phosphate synthase (MIPS), encoded by the ISYNA1 gene (see below). The crucial role of inositol-containing compounds in cell function is underscored by the link between perturbation of inositol metabolism and human disorders, including bipolar disorder, Alzheimer’s and Parkinson’s diseases, polycystic ovary syndrome, diabetes, and various cancers. Therefore, elucidating the mechanisms underlying the control of inositol homeostasis is expected to have important implications for a broad range of illnesses.

Given the vital cellular and clinical importance of inositol, it is striking that very little is known about the regulation of inositol homeostasis in human cells. The long-term goal of our research is to elucidate the molecular mechanisms underlying regulation of inositol homeostasis, to identify the cellular consequences of inositol deprivation, and to determine the therapeutic mechanisms of drugs used to treat bipolar disorder, including VPA and Li, which have been shown to inhibit the inositol biosynthetic pathway. Using relevant yeast and mammalian cell lines (including our unique ISYNA1-KO human cell line and inositol-free culture system), we are addressing the following questions:
- What is the mechanism by which IP6K1 represses ISYNA1 expression?
- Does inositol deprivation activate the unfolded protein response by upregulating ceramide?
- How does inositol deprivation lead to perturbation of mitochondrial function?
- What are the cellular consequences of mood stabilizer treatment?